
Regulatory Considerations for Medical Device Software
In the first installment in ICS’ series What You Need to Know About Developing SaMD we provided an overview of software used within a healthcare environment and touched on key regulations in the U.S and in the EU. In this article, we take a closer look at some of the regulatory considerations for medical device software, including software classification, regulatory pathways to market, and key SaMD documentation.
Classifying Medical Device Software
Properly classifying your SaMD is crucial as it determines your medical device path to the market and requirements that it should meet. Classification differs between regions, with the FDA and EU offering differing perspectives.
FDA Perspective
In the U.S., the medical device classification is based mainly on the risk posed by the medical device. Classifications include: class I, class II, and class III. To determine your medical device class you need to know both regulation number and product code. To find this you should review the categories established by the FDA and identify the one most in line with your intended use and indications for use.
Next, you should find the product code applicable to your medical device. If you determine that your medical device is class I, the general control applies. Class II requires general and special controls, which usually means that you have to submit 510(k) to the FDA. If your device is class III you should get premarket approval (PMA) before launching your product.
There’s also a DeNovo certification pathway dedicated to novel devices for which there is no legally marketed predicate device.
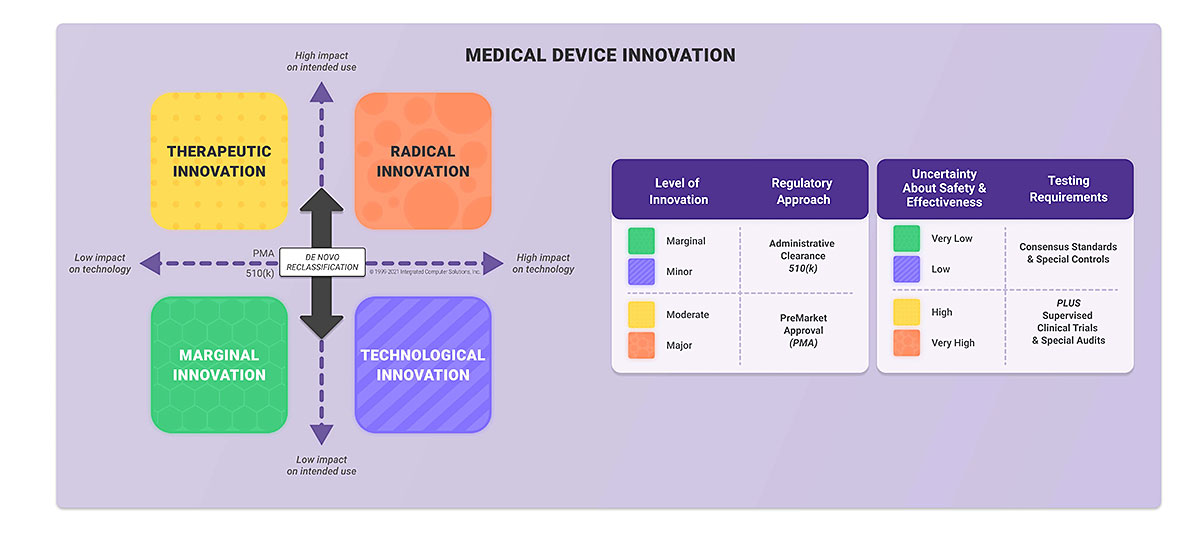
There is one more concept of classification developed by the FDA. This is called the Level of Concern (LOC). It influences the extent of documentation you need to submit to the FDA and it is not related to the device classification described above. There are three LOC: Minor, Moderate and Major. You can determine your level by answering questions provided in the FDA Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices.
Note that in the latest version of this guidance, drafted in November 2021, the LOC has been replaced by four risk-based factors that determine whether you should submit Basic Documentation or Enhanced Documentation. You should submit the latter if any of the following factors apply:
- The software is a constituent part of a combination product;
- The device is intended to test blood donations for transfusion-transmitted infections, or is used to determine donor and recipient compatibility, or is a Blood Establishment Computer Software;
- The device is classified as class III; and/or
- A failure or latent flaw of the device software function could present a probable risk of death or serious injury, either to a patient, user of the device, or others in the environment of use. These risks should be assessed prior to implementation of risk control measures.
EU Perspective
The EU classification is similar to that of the FDA and encompasses class I (with measuring function, sterile), class IIa, class IIb, and class III. The class of your medical device depends mainly on the duration of use, invasiveness and whether your device is active. The rules are outlined in the EU MDR 2017/745 Annex VIII.
For medical device software, Rule Number 11 is applicable. According to this rule, the medical device software can be classified as a medical device class I, IIa, IIb or III depending on the intended use and severity of the potential harm that can be caused by the software.
If your medical device is class I you can self-certify, i.e. prepare required documentation and submit to the relevant entity in your country. If your medical device is higher class or has a measure function or is sterile, the Notify body needs to take part in the conformity assessment and issue a CE certificate.
62304 Perspective
To complicate matters, there is an additional classification outlined in the 62304 standard. This classification determines the 62304 requirements that your software should meet. According to this standard, you should assign one of the following safety classification for each software system:
Class A — If your software can’t contribute to a hazardous situation or can contribute to a hazardous situation that does not result in unacceptable risk after implementing risk-control measures.
Class B — If your software can contribute to a hazardous situation that results in unacceptable risk after consideration of risk-control measures external to the software and the resulting possible harm is non-serious injury.
Class C — If your software can contribute to a hazardous situation that results in unacceptable risk after consideration of risk-control measures external to the software system and the resulting possible harm is death or serious injury.
Documentation
Developing medical device software means documenting nearly everything you do. The content of the medical device file depends on the classification described in the section above. The most common documents are:
Software Development Plan (SDP) — description of software life cycle processes including design and development, configuration management, change management, and maintenance.
Device Hazard Analysis — tabular description of software hazards, their cause, severity and mitigations.
Software Architecture — description and diagrams of the software structure, modules, layers, and interfaces.
Software Requirements Specification (SRS) — description of all software functions and capabilities.
Software Detailed Design (SDD) — specification of software components design, interfaces and responsibilities.
Verification and Validation — description of the testing at the unit, integration and system level including test protocols, expected results, observations and summary.
Traceability — traceability of requirements, specifications, identified hazards and mitigations, and performed testing.
Release Notes — revision history log and list of all known residual software anomalies.
Summary
The sheer volume of regulatory requirements to apply, guidance to follow, and documents to write can seem overwhelming. However, the earlier in the software development process you start thinking about compliance, the easier it is to meet the requirements and ultimately provide high-quality and safe software products.
In the next installment in our SaMD series, we explore mobile medical apps and identify key aspects to consider when designing and developing medical device software intended for use on smartphones, tablets and other mobile platforms. Missed part 1? Read What You Need to Know About Developing SaMD.